
La regolamentazione del brevetto farmaceutico: dal 1939 ad oggi
Come è noto il brevetto nacque, in ambito prevalentemente meccanico, al fine di garantire un riconoscimento agli inventori per i loro prodotti e quindi per sollecitare il progresso, scopo che ancor oggi persegue. Successivamente la sua tutela è stata gradualmente estesa sino a permettere la protezione di tutte le possibili invenzioni umane in ambito tecnico; l’applicazione, però, di tale istituto nei diversi campi è stata talvolta travagliata a causa di timori di derive illecite in quanto contrarie alla morale e l’etica pubblica.
Basti pensare che il brevetto in campo biotecnologico è stato regolamentato solo pochi anni fa; questo, infatti, era stato ostacolato da resistenze di ordine morale, che ancor oggi ne limitano l’applicazione. Un percorso altresì accidentato è stato seguito dalla concessione della tutela brevettuale in campo farmaceutico.
Così come oggi la paura di derive amoralistiche portano parte della dottrina ad ostacolare la possibilità di brevettare prodotti biotecnologici, il timore di agevolare “ciarlatani, speziali e segretisti” e che vi fosse un “rincarimento cagionato dalla privativa” fece si che la prima norma italiana sul brevetto di medicamenti, il Decreto Regio n. 1127 del 1939, fosse limitativa; difatti il comma 1 dell’art. 14 vietò in toto la tutela di medicamenti. Tale norma era in linea con l’orientamento dell’epoca a livello europeo, nello specifico il decreto riprendeva la legge piemontese n. 782 del 12 marzo 1855.
È stata la Corte di Cassazione con la sentenza n. 20 del 9 marzo 1978[1] a segnare il cambio di orientamento in Italia, dichiarando l’incostituzionalità del primo comma dell’art. 14 e quindi aprendo la strada alla creazione del brevetto in campo farmaceutico.
Il fermento in tale materia era vivo da tempo: molto Paesi, come la Germania, già avevano previsto la possibilità di brevettare i medicamenti, indice ne è il fatto che la Corte venne adita dalla Commissione Ricorsi la quale, tramite l’opposizione di numerose ordinanze reiteranti il divieto di brevettazione in campo farmaceutico, sollevò la questione di legittimità. Nel proprio provvedimento la Suprema Corte valutò che le motivazioni che spinsero la Commissione parlamentare ad escludere i prodotti farmaceutici dalla brevettabilità non risultavano più attuali: gli arcaici metodi di produzione e sviamento della pubblica opinione erano stati superati con l’industrializzazione e l’introduzione dei mezzi di comunicazione di massa[2]; i prodotti ora, come allora, sono determinati de imperioda leggi sanitarie e dalla normativa del Comitato interministeriale dei prezzi; infine, la possibile rarefazione dei prodotti farmaceutici derivante dalla brevettazione, sarebbe stata, in ogni caso, contrastata dalla stessa legge sul brevetto[3].
La Corte non rinvenne solo il superamento degli ostacoli precedenti ma bensì constatò che: il 1° comma violava l’art. 3 della Costituzione “per l’ingiustificato sacrificio di diritti anche di particolare valore morale che determinerebbe”; l’art. 9 in quanto il brevetto garantisce anche un’incentivazione patrimoniale ed l’impossibilità d’applicarlo in tale materia “intralcerebbe la ricerca scientifica e tecnica nel campo dei medicamenti, dissuadendo l’industria farmaceutica dall’effettuarvi i necessari investimenti”; gli artt. 41, 42 e 43 in quanto oltre a negare i diritti patrimoniali, impedivano l’attribuzione dell’invenzione all’ideatore, o agli aventi diritto, relegandoli, in contrasto con la legge superiore, nell’oblio e lasciando il settore della ricerca non regolato, sottratto all’iniziativa privata, ma non demandato a quella pubblica.
Considerato quindi che la disciplina derogativa non era congrua rispetto all’interesse generale derivante dalla ricerca in campo farmaceutico la Corte Costituzionale ha dichiarato l’illegittimità costituzionale del 1° comma dell’art.14 del d.r. 1127/1939 “Testo delle disposizioni legislative in materia di brevetti per invenzioni industriali”
La decisione è stata recepita con il decreto del Presidente della Repubblica n. 338 del 22 giugno 1979 nel quale, però, in contrasto con quanto suggerito dagli Ermellini, il legislatore si è limitato a rielaborare il testo dell’art.14 senza assicurare una tutela transitoria agli inventori che non avevano potuto brevettare i propri prodotti.
Il Decreto regio così rielaborato stabilì per le invenzioni farmaceutiche, in conformità con la durata brevetto in altri campi, una tutela brevettuale di 20 anni; in tale materia, però, questo termine è risultato essere insufficiente. Spesso, infatti, al fine di ottenere la protezione statale le case farmaceutiche preferivano, ed oggi ancor di più, brevettare il principio attivo; ciò garantiva da una parte di anticipare il deposito e dall’altra di ottenere una maggior ampiezza nella garanzia brevettuale. La nuova normativa, infatti, affinché fosse concesso il brevetto richiedeva solamente che il richiedente avesse il principio attivo, ne conoscesse la funzione e ne dimostrasse, anche solo con simulazioni, almeno una modalità di somministrazione, la quale però non sarebbe stata vincolante per il prodotto finale.
La richiesta della tutela, compiuta in una fase tanto embrionale, comportava, d’altra pare, che a seguito della domanda di brevettazione, la società dovesse ancora sostenere gran parte della sperimentazione clinica con notevole impegno economico e temporale[4]. Inoltre, conclusa la fase sperimentale, prima che un farmaco potesse essere messo in commercio doveva, come oggi, ottenere l’Autorizzazione per l’Immissione in Commercio con la conseguenza che la società dovesse sostenere un’ulteriore spendita dell’arco temporale tutelato senza che la tutela fruttasse i vantaggi economici a cui era preposta.
Al fine di redimere la suesposta questione che comportava l’inadeguato sfruttamento del monopolio garantito il legislatore emanò la legge n. 349 del 19 ottobre 1991 con cui introdusse il Certificato di Protezione Complementare.
Attraverso il CPC l’ideatore o il suo avente causa potevano recuperare il tempo impiegato, per un massimo di 18 anni, nella sperimentazione al momento dell’esaurimento della tutela ventennale secondo il seguente schema:
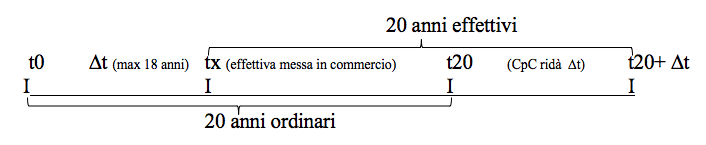
Dove t0 indica il momento in cui viene inoltrata la domanda di brevetto e quindi quando comincia la tutela brevettuale; Dt il tempo impiegato nella sperimentazione clinica e l’ottenimento dell’autorizzazione per la messa in commercio; tx il momento dell’effettiva messa in commercio del farmaco e quindi l’inizio del suo sfruttamento commerciale; t20 il momento della caducazione ordinaria del brevetto (20 anni dalla domanda); t20+Dt il momento della reale caducazione del brevetto grazie alla proroga garantita dal Certificato di Protezione Complementare.
Se, per esempio, a seguito della domanda si fossero impiegati ulteriori 11 anni nella sperimentazione quest’arco temporale sarebbe stato recuperato alla fine dei canonici 20 anni di fatto aumentando la tutela a 31 anni.
La prima importante riforma della materia a livello comunitario avvenne l’anno successivo per mezzo della Comunità Economica Europea che ha disciplinato la brevettazione in campo farmaceutico con il regolamento n. 1768 del 18 giugno 1992, entrato in vigore nel 2 gennaio 1993 e recepito in Italia il 1 gennaio 1994, attraverso cui ha introdotto il Certificato di Protezione Supplementare, ancor oggi in vigore. Il nuovo Certificato permette di prorogare il monopolio brevettuale per un massimo di cinque anni secondo il seguente schema:

Per calcolare, quindi, l’estensione del CPS è necessario considerare gli anni di effettiva sperimentazione (Dt) a cui vanno sottratti 5 anni, per tale motivo è concedibile solo se tra la domanda di brevetto e la commercializzazione intercorrono almeno 5 anni; il nuovo istituto, inoltre, prevede una proroga massima di 5 anni[5].
Riprendendo l’esempio fatto precedentemente nel caso di una sperimentazione di 11 anni non si avrà più una tutela totale di 31 anni bensì di 25.
Da tale schema si deduce che con l’entrata in vigore della normativa europea si è assistito ad una riduzione effettiva della tutela da 20 a 15 anni[6].
La normativa italiana in tale campo ha compiuto un ultimo intervento legislativo con la legge n. 112 del 15 giugno 2002, teso a riallineare la durata del Certificato di Protezione Complementare con quello introdotto a livello europeo. Quest’ultima modifica nel panorama normativo del brevetto farmaceutico è stata contestuale alla scadenza dei primi brevetti farmaceutici concessi, con la caduta in pubblico dominio di questi farmaci all’originator si sono affiancati numerosi farmaci generici dal costo notevolmente inferiore. Si ricordi che il Sistema Sanitario Nazionale sostiene il costo dei farmaci di fascia A coprendo il prezzo più basso per ciascun farmaco, per cui, con l’entrata in commercio dei farmaci generici – prodotti di uguale composizione, forma farmaceutica, via di somministrazione, modalità di rilascio e indicazioni terapeutiche[7] – il SSN ha avuto un notevole risparmio. Con la legge del 2002 il legislatore ha modificato i termini dei Certificati di Protezione Complementare emessi restituendo un anno di protezione monopolistica per ogni anno e mezzo di sperimentazione per cui permettendo di recuperare non più la totalità degli anni impiegati nella sperimentazione ma solo i 2/3. Per mezzo di quest’ultima regolamentazione ad oggi sono scaduti tutti i Certificati di Protezione Supplementare ed il brevetto farmaceutico può essere accompagnato solamente dal Certificato di Protezione Complementare previsto a livello unitario.
Tale istituto è riuscito a contemperare l’esigenza d’ampliamento della tutela richiesta dalle imprese farmaceutiche con le esigenze economiche avanzate dai sistemi sanitari tanto che nel 2009 il legislatore europeo ha emesso un nuovo regolamento[8] che ha abrogato e sostituito il precedente regolamento comunitario, 1768 del 1992, sostanzialmente riproponendo l’istituto del certificato protettivo complementare così come regolamentato in precedenza.
[1] La Corte era stata adita dalla Commissione Ricorsi contro i provvedimenti dell’Ufficio centrale brevetti. L’attrice era stata a sua volta adita separatamente da ditte di varia nazionalità: Ciba Societé anonyme, Dr. Madaus & Co., Lovens Kemiske Productionsaktienselkab, Rohm & Haas G.m.b.H., Smith Kline & French Laboratories, Ugine Kuhlmann, due volte ricorrente, The Upjohn Company, United States Borax and Chemical Corporation, Beecham Group Limited, Farbwerke Hoechst Aktiengesellschaft, due volte ricorrente, Kyowa Hakko Kogyo Co. Ltd., Fisons Pharmaceutical Ltd., RhonePoulenc s.a., I.S.F. s.p.a., due volte ricorrente, Astra Pharmaceutical Products Inc.
[2] La Corte sottolineò inoltre che la norma regia era parziale, in quanto rinviava la disciplina della materia ad un “codice sanitario ed igienico” che si stava improntando in quel periodo, ma che non è mai stato emanato.
[3] L’art. 52 della legge sul brevetto vigente nel 1978, attuale art.69 del Cpi, prevedeva un onere d’attuazione nel territorio dello Stato in misura tale da non risultare in grave sproporzione con i bisogni del paese.
[4] La sperimentazione clinica è composta da quattro fasi: la Cinestesi, che prevede la somministrazione del principio attivo in un insieme di uomini sani; gli studi di gruppo, che prevedono la somministrazione ad un gruppo più ampio di soggetti; gli studi su un campione di pazienti già affetti da patologie o condizioni morbose; farmacovigilanza, che prevede la messa in commercio del farmaco con la supervisione da parte dell’Aifa.
[5] La Corte di Giustizia Europea ha stabilito che, dovendo sempre sottrarre 5 anni alla perdita reale, il certificato di protezione supplementare può anche essere negativo.
[6] La riduzione della tutela garantita dal 1994 in poi causò, nei mesi precedenti, una corsa al brevetto; l’art. 20 del regolamento 1768 del 1992, infatti, lasciò in vigore i certificati nazionali richiesti prima del recepimento del CPC o comunque collegati ad una domanda di brevetto depositate prima di tale data.
[7] Definizione ripresa dal ministero della salute, disponibile qui: http://www.salute.gov.it/portale/esenzioni/dettaglioContenutiEsenzioni.jsp?lingua=italiano&id=4674&area=esenzioni&menu=vuoto
[8] Regolamento (CE) n. 469/2009 del Parlamento Europeo e del Consiglio del 6 maggio 2009 sul certificato protettivo complementare per i medicinali, disponibile qui:
Fonti normative:
Decreto Regio n. 1127 del 1939
Sentenza della Corte Costituzionale n. 20, 9 marzo 1978
Decreto del Presidente della Repubblica n. 338 del 22 giugno 1979
Legge n. 349 del 19 ottobre 1991
Regolamento n. 1768 del 18 giugno 1992
Legge n. 112 del 15 giugno 2002
Regolamento n. 469 del 6 maggio 2009
Bibliografia:
Ghidini, Profili evolutivi del diritto industriale, 2015.
M. Di Condojanni, Abuso del diritto e proprietà industriale: il mercato farmaceutico, 2018
Nicoletta Cosa si è laureata in Giurisprudenza presso La Sapienza Università di Roma nel novembre 2017. Sta proseguendo gli studi partecipando al Master in diritto della Concorrenza ed Innovazione presso la Luiss School of Law. Attualmente è anche praticante presso un prestigioso studio legale della capitale.



Pingback: Menarini e la ricerca farmaceutica - Made in Italy